

Система учета научной деятельности (ASSA) |


|
Лаборатория компьютерной протеомики (т.12)Отдел системной биологии
Научные результаты Сотрудники О Подразделении 1. Основное направление исследований Изучение структурно-функциональной организации белков
2. Задачи, решаемые в рамках базового бюджетного проекта на данном этапе: В рамках проекта VI.61.1.2. «Компьютерно-экспериментальное исследование и моделирование структурно-функциональной организации и эволюции генных сетей многоклеточных и одноклеточных организмов» решаются задачи по направлению компьютерный анализ и моделирование структурно-функциональной организации белков, включая: (1) совершенствование существующих и разработка новых алгоритмов компьютерной структурно-функциональной аннотации белков (методы молекулярного моделирования, методы предсказания сайтов связывания малых химических соединений, сайтов белок-белок взаимодействий, активных центров и методы предсказания термодинамической стабильности белков); (2) исследование взаимосвязи между структурой белков и их функциональными, аллергенными и иммуногенными свойствами на основе анализа конформационных пептидов, моделирующих белковые молекулярные поверхности; (3) изучение закономерностей структурной организации функциональных сайтов белков; (4) анализ закономерностей структурной организации ассоциативных сетей белок-белок взаимодействий, связанных с заданной клеточной/системной функцией, биологическим процессом или заболеванием; (5) разработка новых подходов для поиска фармакологических мишеней и дизайна молекулярных соединений – кандидатов для лекарственных препаратов и проведение, на этой основе, компьютерного дизайна/скрининга ряда химических соединений, обладающих потенциальным противовирусным и антибактериальным действием; (6) изучение влияния мутаций на изменение термостабильности белков, функционирующих в составе изучаемых генных сетей, с использованием методов молекулярной динамики. (7) создание интегрированных информационных хранилищ данных по структуре, функции и эволюции белков. С помощью интегрированных подходов будет проводиться анализ метапротеомов бактериальных сообществ.
3. Прикладные разработки Разработана программно-информационная система Protein Structure Discovery (Иванисенко и др., 2011), предназначенная для решения широкого круга задач компьютерной протеомики, включая предсказание функции, структуры и иммунологических свойств белков. Специально созданный раздел системы позволяет оценивать количественные и качественные эффекты мутаций на структурные и функциональные свойства белков. Всего в систему интегрировано более 20 различных программ, в том числе база данных PDBSite (Ivanisenko et al, 2005), содержащая информацию о пространственных структурах более 100000 функциональных сайтов белков, программа распознования функциональных сайтов в пространственных структурах белков PDBSiteScan (Ivanisenko et al, 2004), программа количественного анализа взаимосвязи структура-активность белков WebProAnalyst (Ivanisenko et al, 2005), система SitEx (Medvedeva et, al, 2012), предназначенная для решения задач по анализу соотношения экзон-интронной структуры генов и структурно-функциональной организации, кодируемых ими белков; программа Allpred (Bragin et al, 2013), предназначенная для предсказания аллергенности белков с использованием конформационных пептидов и другие. Protein Structure Discovery имеет Вэб-интерфейс и доступна пользователям через Интернет (http://www-bionet.sscc.ru/psd).
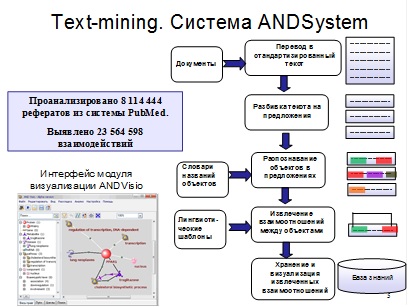
4. Иллюстрированное описание лучших результатов, полученных подразделением за последние 5 лет. Общий объем текста на одну лабораторию (сектор) – не более 4000 знаков с пробелами, количество иллюстраций с подписями: от одной до – пяти. Рекомендуется, чтобы общий объем текста с иллюстрациями не занимал более 5 страниц в формате Word (для текста шрифт Nimes New Roman, фонт № 12, через 1 интервал). 1) Разработана компьютерная система ANDSystem автоматического анализа текстов научных публикаций и баз данных (Demenkov et al, 2011). Система содержит модуль извлечения знаний из текстов и баз данных, базу знаний ANDCell и программу ANDVisio, обеспечивающую доступ к базе знаний и визуализацию ассоциативных генных сетей (рис. 1). Система позволяет автоматически извлекать из текстов информацию о широком круге взаимодействий и взаимоотношений между белками, генами, микроРНК, метаболитами, болезнями и биологическими процессами, включая физические взаимодействия, регуляторные отношения, транспорт и др.
Рисунок 1. Схематичное представление системы ANDSystem автоматического извлечения знаний из текстов научных публикаций и биологических баз данных. 2) Найден гипотетический рецепторный домен служащий рецептором для проникновения прионов и вирусов в клетку (Малыгин и др. 2009). Проведено компьютерное моделирование пространственной структуры ламининсвязывающего белка человека (LBP). Показано, что разрывные участки последовательности C-концевого домена LBP, взаимодействующие с гликопротеином Е вируса венесуэльского энцефалита лошадей (VEE) и вируса клещевого энцефалита (TBE), оказываются сближенными в пространственной структуре и образуют рецепторный домен. Кроме того, анализ показал, что данный домен служит рецептором для проникновения прионов в клетку, таким образом, существует общий путь проникновения для вирусов и прионов (рис. 2).
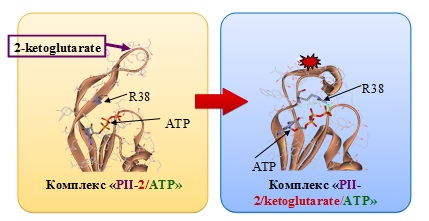
3) Компьютерный анализ белка PII Mycobacterium tuberculosis, являющегося важнейшим регулятором метаболизма азота, сигнальной трансдукции и мембранного транспорта в микобактерии, проведенный с использованием программы PDBSiteScan и молекулярной динамики, выявил в данном белке сайты связывания 2-кетоглютората (рис. 3). Молекулярное моделирование показало, что связывание 2-кетоглютората приводит к перестройке белка PII и повышению его сродства к АТФ. Экспериментальная проверка на приборе BIOCORE подтвердила результаты компьютерного анализа (Bandyopadhyay et al., The Journal of Biochemistry, 2010). Таким образом, показано, что 2-кетоглюторат действует как аллостерический регулятор связывания PII с ATФ.
Рисунок 3. Графическое изображение фрагмента модели пространственной структуры белка PII в комплексе с АТФ и 2-кетоглютарат.
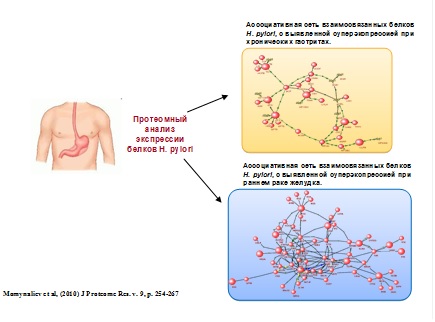
4) На основе анализа экспериментальных данных протеомных исследований, полученных в Институте физико-химической медицины (Москва) проведена реконструкция и анализ сетей молекулярно-генетических взаимодействий белков Helicobacter pylori, дифференциально экспрессирующихся в различных штаммах, выделенных у пациентов с хроническими гастритами и опухолями желудка (рис. 4). Показано, что различия в экспрессии белков H. pylori могут быть связаны с адаптацией этого микроорганизма к различным условиям внешней среды, обусловленными морфологическими отличиями различных участков желудка, подверженных гистологическим и морфологическим изменениям в ходе патологических процессов (Momynaliev et al, 2010). Для реконструкции ассоциативных сетей использовалась система автоматического анализа текстов научных публикаций и баз данных ANDSystem.
Рисунок 4. Ассоциативные сети взаимосвязанных белков H. pylori, идентифицированных в протеомных экспериментах, построенные с использованием системы автоматического анализа научных текстов ANDSystem.
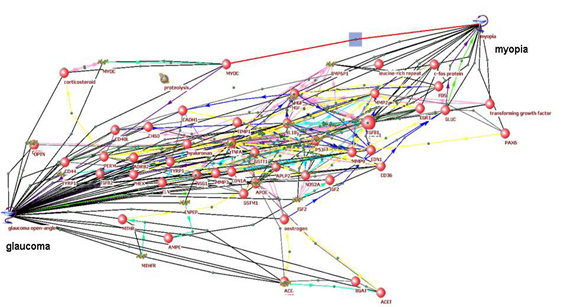
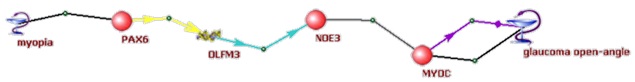
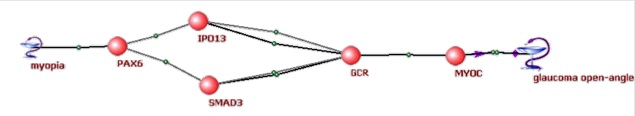
5) С использованием компьютерной системы ANDSystem проведена реконструкция и анализ ассоциативной сети, описывающей потенциальные молекулярные механизмы взаимосвязи миопии и открытоугольной глаукомы (Подколодная и др., 2010). Показано, что ассоциативная сеть содержит более 200 различных генов, ассоциированных с этими заболеваниями, и более чем 2000 молекулярно-генетических взаимодействий между данными генами. Редукция сети и дальнейший анализ представленных в ней молекулярно-генетических путей позволили выявить ряд потенциальных генов-кандидатов для генотипирования одновременно миопии и открытоугольной глаукомы (рис. 5).
А
Б
В Рисунок 5. Молекулярно-генетические пути, ассоциированные с открытоугольной глаукомой и миопией. (А) Наиболее значимые взаимодействия между генами и белками человека, ассоциированными с открытоугольной глаукомой и миопией; (Б) пути, направленные от миопии к глаукоме; (В) пути, с нечетко определенным типом и направлением взаимодействий.
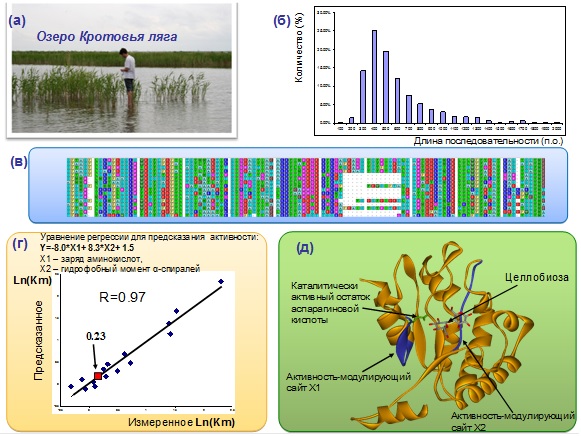
6) Разработан (совместно с Институтом физико-химической биологии ФМБА) новый подход к поиску белков, перспективных для биотехнологии (Иванисенко и др., 2012). Подход основан на параллельном высокопроизводительном секвенировании метагеномов и компьютерном анализе: сборке коротких секвенированных фрагментов в протяженные геномные последовательности (контиги); поиске в них открытых рамок считывания; функциональной классификации кодируемых ими ферментов и предсказании количественных величин их функциональной активности. На основе анализа результатов секвенирования метагенома микробных сообществ озера «Кротовья ляга», выполненного на платформе SOLiD, идентифицирован белок гликозидаза (фермент деградации целлюлозы) с высокой функциональной активностью (рис. 6).
Рисунок 6. Новый подход к поиску белков, перспективных для биотехнологии, основанный на параллельном высокопроизводительном секвенировании метагеномов и компьютерном анализе. (а) Озеро «Кротовья ляга» - источник генетического материала для метагеномного секвенирования микробных сообществ; (б) распределение длин нуклеотидных последовательностей, собранных в контиги; (в) фрагмент множественного выравнивания белковых последовательностей, потенциальных гликозидаз, кодируемых открытыми рамками считывания; (г) предсказание значения количественной величины константы Михаэлиса для потенциальной гликозидазы в реакции расщепления целлобиозы; (д) реконструированная модель пространственной структуры гликозидазы, обладающей повышенной активностью расщепления целлобиозы.
7) Предсказана мишень действия соединения NIH415032 (рис. 7), обладающего согласно экспериментальным данным противотуберкулезной активностью (Gahoi et al, 2013). С использованием программы PDBSiteScan и методов виртуального скрининга показано, что миколилтрансфераза (антиген 85) M. tuberculosis, один из ключевых ферментов, участвующих в биосинтезе микобатериальных клеточных стенок, является потенциальной мишенью для соединения NIH415032, для которого ранее было показана противотуберкулезная активность, но мишень его действия была не известна (Gahoi et al, 2013).
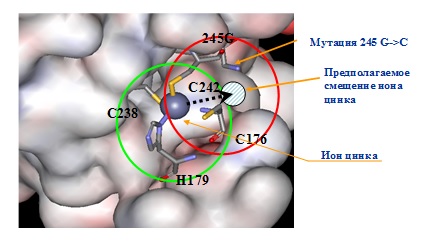
8) С помощью программы предсказания функциональных сайтов в пространственных структур белков PDBSiteScan и методов молекулярной динамики изучен структурный эффект действия мутации Gly245-Сys в ДНК-связывающем домене белка p53, ассоциированной с наследственной предрасположенностью к раку (синдром Ли-Фраумени). Показано, что мутация приводит к появлению нового сайта связывания иона цинка в непосредственной близости с существующим цинк-связывающим сайтом (рис. 8). Смещение иона цинка в данный сайт ведет к существенным конформационным перестройкам белка, что может обуславливать нарушения его функций (Pintus et al, 2013).
Рисунок 8. Графическое изображение фрагмента пространственной структуры ДНК-связывающего домена белка p53, в котором согласно расчетам в результате мутации Gly245-Сys возникает новый цинк-связывающий сайт.
5. Задачи, планируемые на перспективу: формулировки должны содержать как фундаментальные научные задачи, так и возможную прикладную направленность исследований в подразделении. Белки представляют собой важнейшую компоненту ГС. Наряду со структурными, каталитическими, транспортными функциями, белки в ГС выполняют важнейшие регуляторные функции: контролируют экспрессию генов, обеспечивают формирование сетей белок-белковых, РНК/ДНК-белковых и белок-лигандных регуляторных взаимодействий, путей передачи сигналов от клеточных мембран к ядрам клеток, межклеточных коммуникаций. Функциональная активность белков в ГС определяется наборами характерных для них функциональных сайтов. По данным базы PDB (Rose et al., 2011) в расшифрованных пространственных структурах белков может содержаться от нескольких до десяти и более функциональных сайтов. Однако, база данных PDB содержит в настоящее время описание 3-D структур только для ~ 80 000 белков. Из них уникальные пространственные структуры составляют ~ 15%, а остальные являются изоформами или мутантными формами. При этом в PDB представлено около 20 000 уникальных 3-D структур белков человека, имеющих гомологию менее 95%. Таким образом, до сих пор имеется большой разрыв между информацией, представленной в базе данных и полным количеством белков, кодируемых геномом человека с учетом вариантов альтернативного сплайсинга, что затрудняет оценку влияния генетической изменчивости на функциональные сайты белков. Поэтому важнейшее значение имеет создание высокопроизводительных конвейерных методов компьютерного предсказания функциональных сайтов в пространственных структурах белков. В связи с этим планируется решение следующих задач из области компьютерного анализа структурно-функциональной организации белков, функционирующих в биомедицински значимых ГС человека: предсказание компьютерными методами функциональных сайтов в пространственных структурах этих белков; оценка на этой основе репертуаров белок-белковых, РНК/ДНК-белковых и метаболит-белковых взаимодействий, и реконструкция сетей межмолекулярных взаимодействий, значимых для функционирования изучаемых ГС; картирование аминокислотных полиморфизмов на функциональные сайты белков и изучение на этой основе влияния мутаций на структуру и функцию белков, а также на формируемые ими сети межмолекулярных взаимодействий; изучение влияния мутаций на термодинамическую стабильность и специфическую активность белков, а также на структурные и конформационные свойства функциональных сайтов в трехмерной структуре белков. Короткие природные пептиды обладают широкой биологической активностью, включая регуляцию деятельности высшей нервной системы. Механизмы их действия до сих пор остаются слабо изученными. Наша идея состоит в том, что пептиды могут взаимодействовать с рецепторами за счет структурной мимикрии функциональных сайтов белков, для которых эти рецепторы являются мишенями. Будет создана база данных выравниваний последовательностей коротких пептидов с конформационными пептидами белков, характеризующая потенциальные молекулярные функции коротких пептидов и их мишени по аналогии с белками, имеющими сходство с этими пептидами. Будет создан параллельный алгоритм для предсказания функциональных сайтов в 3-D структурах белков на основе метода PDBSiteScan. Будут предсказаны функциональные сайты в пространственных структурах белков, функционирующих в составе изучаемых ГС, включая сайты, возникающие в результате генетической изменчивости, с помощью новой версии программы. Особое внимание будет обращено на функциональные сайты белков, выполняющих регуляторные функции (сайты аллостерической регуляции белков; сайты связывания РНК/ДНК, сайты, участвующее в формировании сетей белок-белковых взаимодействий). Будет создана реляционная версия базы данных PDBSite, интегрирующая информацию по экспериментально установленным и предсказанным (потенциальным) сайтам в 3-D структуре белков. Будет проведена классификация сайтов по сходству их пространственных структур, физико-химических и конформационных свойств, создание обобщенных шаблонов, описывающих различные структурные и функциональные классы сайтов. Будет осуществлена интеграция PDBSite с внешними базами данных Gene Ontology (GO), UniProt, PDB и др. Будет осуществлено расширение базы данных PDBSite: картирование микроэволюционной (популяционной) и макроэволюционной генетической изменчивости на пространственные структуры и функциональные сайты белков, функционирующих в составе изучаемых ГС, включая: - популяционные полиморфизмы из баз данных "1000 геномов", dbSNP, OMIM и др. - нуклеотидные и аминокислотные замены, выявляемые во множественных выравниваниях семейств белков, функционирующих в составе изучаемых ГС; - особое внимание будет обращено на аминокислотные замены в белках, выполняющих в исследуемых ГС регуляторные функции, а также ассоциированных с социально значимыми заболеваниями человека. Будет осуществлено развитие методов молекулярного моделирования динамики и механики белков для анализа функциональных сайтов и их взаимодействия с различными лигандами, включая малые химические соединения, белки, молекулы РНК и регуляторные сайты в ДНК. Будет разработан параллельный алгоритм для оценки изменений термодинамической стабильности мутантных белков на основе методов ProtStability. Будет исследовано влияние аминокислотных замен, представленных в базе данных PDBSite, на термодинамическую стабильность и специфическую активность белков, в том числе белков, функционирующих в составе изучаемых ГС. Будет исследовано влияние аминокислотных замен, представленных в базе данных PDBSite, на структурные и конформационные свойства функциональных сайтов белков, включая сайты, локализованные в белках, функционирующих в составе изучаемых ГС. Будет изучено влияние замен аминокислот на конформационную подвижность сайтов, изменения их физико-химических характеристик, устойчивость сетей водородных связей, формируемых аминокислотными остатками функциональных сайтов и т.д. Будет создан параллельный алгоритм для предсказания величин специфических активностей белков на основе разработанного нами ранее метода WebProAnalyst, использующего в качестве исходной информации множественные выравнивания семейств белков с известными величинами их специфической активности. Будет проведен анализ влияния аминокислотных замен, описанных в базе PDBSite, на специфическую активность белков, включая белки, функционирующие в составе изучаемых ГС. Будет проведена интеграция разработанных информационных и программных продуктов в систему «КОМПЬЮТЕРНАЯ ПРОТЕОМИКА» для конвейерного высокопроизводительного анализа структурно-функциональной организации белков и их функциональных сайтов, обеспечивающего: - предсказание пространственной (3-D) структуры белков по их аминокислотным последовательностям; - быстрый поиск структурной гомологии белков по базе данных PDB; - предсказание функциональных сайтов в 3-D структурах белков; - количественный анализ взаимосвязи «структура-активность» в семействах родственных и мутантных белков; - предсказание изменения термодинамической стабильности мутантных белков; - молекулярное моделирование динамики и механики белков; - оценку влияния аминокислотных замен в белках на их структурно-функциональные характеристики. Будет создана база знаний по вывяленным особенностям структурно-функциональной организации белков, функционирующих в составе изучаемых ГС, включая влияние мутаций на их структуру и функцию, и расширение на этой основе ГС. Антропова Евгения Александровна [младший научный сотрудник] Деменков Павел Сергеевич [научный сотрудник] Иванисенко Тимофей Владимирович [младший научный сотрудник] Клещев Максим Александрович [научный сотрудник] Твердохлеб Наталья Николаевна [программист 1 категории] Яцык Иван Викторович [инженер-исследователь] Бывшие сотрудникиАлемасов Николай АлександровичАнарбаев Рашид Октамович Жамбалдагбаев Нима Цыренович Завьялов Владимир Юрьевич Иванисенко Никита Владимирович Медведева Ирина Вадимовна Мищенко Елена Леонидовна Петровский Евгений Дмитриевич Сайк Ольга Владимировна Степаненко Ирина Лембитовна СовместителиВолянская Анастасия Рашидовна [младший научный сотрудник]Выберите слайдером нужный промежуток, и список ниже будет содержать записи только нужного периода: 97 98 99 00 01 02 03 04 05 06 07 08 09 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 Публикации Конференции Гранты Научное руководство Патенты
|

| 2011 | Protein Structure Discovery: пакет программ для решения задач компьютерной протеомики Иванисенко В. А., Деменков П. С., Иванисенко Т. В., Колчанов Н. А. Биоорганическая химия, 2011, 2011, Т. 37, № 1, С. 22–35 |
| Application of the ANDCell Computer System to Reconstruction and Analysis of Associative Networks Describing Potential Relationships between Myopia and Glaucoma O. A. Podkolodnaya, E. E. Yarkova, P. S. Demenkov, O. S. Konovalova, V. A. Ivanisenko, and N. A. Kolchanov Russian Journal of Genetics: Applied Research, 2011, 2011, Vol. 1, No. 1, pp. 21–28. |
|
| Извлечение знаний из текстов научных публикаций и создание баз знаний в области нанобиотехнологии В.А. Иванисенко, Н.Л. Подколодный, П.С. Деменков, Т.В. Иванисенко, О.А. Подколодная, Е.В. Игнатьева, Т.М. Хлебодарова, Н.Н. Подколодная, Е.А. Ананько, С.С. Гончаров, Н.А. Колчанов Российские нанотехнологии, 2011, 2011 . Т. 6 № 7- 8, с. 14-21. |
|
| 2010 | Использование компьютерной системы ANDCell для реконструкции и анализа ассоциативных сетей потенциальных механизмов взаимосвязи миопии и глаукомы О.А.Подколодная, Е.Э. Яркова, П.С.Деменков, О.С.Коновалова,. В.А.Иванисенко, Н.А.Колчанов Информационный вестник ВОГИС, 2010, Т. 14, № 1. — С. 106–115. |
| Качество спермы и уровни репродуктивных гормонов у мужчин г. Кемерово Н. В. Гуторова, Л. В. Осадчук, М. А. Клещев, Н. Н. Кузнецова, А. В. Осадчук Проблемы репродукции, 2010, №6, стр. 89-93 |
|
| Характеристика C-концевой части гликопротеина Е вируса Западного Нила и оценка силы его взаимодействия с V 3 интегрином как с предполагаемым клеточным рецептором М. В. Богачек, Б. Н. Зайцев, S. K. Sekatskii, Е. В. Протопопова, В. А. Терновой, А. В. Иванова, А. В. Качко, В. А. Иванисенко, G. Dietler, В. Б. Локтев. BIOCHEMISTRY-MOSCOW+, 2010, том 75, вып. 4, с. 571 – 581 |
|
| Экспериментальная проверка функциональной активности потенциальных DRE-сайтов, обнаруженных в промоторах IRF1, REL и IL12A генов человека Е.В. Кашина, Е.В. Антонцева, М.Ю. Шаманина, Е.А. Ощепкова, Д.Ю. Ощепков, А.В. Катохин, А.Ю. Гришанова, Д.П. Фурман, В.А. Мордвинов Труды ТГУ. Серия: Биологическая, 2010, Том 275. С. 353-356. |
|
| Эффективная реализация алгоритма расчёта ближних невалентных взаимодействий на процессоре PowerXCell8i Алемасов Н.А., Фомин Э.С. Вычислительные технологии, 2010, №6, Т. 15, С. 3-18 |
|
| Expression and molecular characterization of the Mycobacterium tuberculosis PII protein Bandyopadhyay A, Arora A, Jain S, Laskar A, Mandal C, Ivanisenko VA, Fomin ES, Pintus SS, Kolchanov NA, Maiti S, Ramachandran S. J BIOCHEM, 2010, 147(2):279-89. |
|
| Functional Divergence of Helicobacter pylori Related to Early Gastric Cancer Momynaliev KT, Kashin SV, Chelysheva VV, Selezneva OV, Demina IA, Serebryakova MV, Alexeev D, Ivanisenko VA, Aman E, Govorun VM. J PROTEOME RES, 2010, 9(1):254-67 |
|
| Visualization and analysis of a cardio vascular disease- and MUPP1-related biological network combining text mining and data warehouse approaches Sommer B, Tiys ES, Kormeier B, Hippe K, Janowski SJ, Ivanisenko TV, Bragin AO, Arrigo P, Demenkov PS, Kochetov AV, Ivanisenko VA, Kolchanov NA, Hofestädt R J INTEGR BIOINFORM, 2010, 7(1):148. doi: 10.2390/biecoll-jib-2010-148 |
|
| Веб-ориентированная система построения и исполнения комплексных запросов к реляционным источникам биологической информации на основе семантической формализации структуры данных Коротков Р.О., Деменков П.С., Иванисенко В.А. Вестник НГУ. Серия: Информационные технологии, 2010, Т. 8. № 3. С. 13-22. |
|
| Высокая частота субоптимального качества спермы у жителей Сибирского региона (на примере г. Новосибирска) Осадчук Л.В.,. Клещев М.А, Темников Н.Д., Еркович А.А., Осадчук А.В. Андрология и генитальная хирургия, 2010, № 3. С. 52-55 |
|
| 2009 | Картирование рецепторного домена для вирусов Венесуэльского энцефалита лошадей и Клещевого энцефалита на С-конце ламининсвязывающего белка человека А.А. Малыгин, Е.И. Бондаренко, В.А. Иванисенко, Е.В. Протопопова, Г.Г. Карпова, В.Б. Локтев BIOCHEMISTRY-MOSCOW+, 2009, т. 74, вып. 12. с. 1631 – 1641 |
| Математическое моделирование действия потенциальных противовирусных препаратов на репликацию субгеномного репликона вируса гепатита С в клетке Е.Л. Мищенко, K.Д. Безматерных, В.А. Иванисенко, В.А. Лихошвай, Н.А. Колчанов Информационный вестник ВОГИС, 2009, №1, Том 13, 208-218 |
|
| Implementation of a Non-bonded Interaction Calculation Algorithm for the Cell Architecture Fomin E.S., Alemasov N.A. Lect Notes Comput Sci, 2009, Vol. 5698, P. 399–405 |
|
| Promoters of the Genes Encoding the Transcription Factors Regulating the Cytokine Gene Expression in Macrophages Contain Putative Binding Sites for Aryl Hydrocarbon Receptor D. P. Furman, E.A. Oshchepkova, D.Yu. Oshchepkov, M.Yu. Shamanina, V.A. Mordvinov COMPUT BIOL CHEM, 2009, V. 33. №6. P. 465-468. |
|
| Анализ распределения аденозин-фосфат связывающих сайтов белков на экзонной структуре гена Медведева И. В., Деменков П. С., Иванисенко В. А. Информационный вестник ВОГИС, 2009, Том 13, №1, стр. 122-127 |
|
| Выявление новых DRE в регуляторной области генов человека, кодирующих компоненты цитозольного комплекса арил-гидрокарбонового рецептора Д.Ю. Ощепков, Д.П. Фурман, Е.А. Ощепкова, А.В. Катохин, М.Ю. Шаманина, В.А. Мордвинов Информационный вестник ВОГИС, 2009, Т.13. № 1. С. 46-52. |
|
| 2008 | Functional divergence of H-pylori related to early gastric cancer Momynaliev KT, Kashin SV, Chelysheva VV, Selezneva OV. Demina IA, Serebryakova MV, Ivanisenko VA, Aman E, Akopian T, Govorun VM. HELICOBACTER, 2008, 5, 13, 477-477 |
| Regulatory region of human genes encoding macrophageal transcription factors possess multiple potential dioxin response elements Oshchepkova EA, Furman DP Oshchepkov DY, Katokhin AV, Shamanina MY, Mordvinov VA, Tsyrlov IB Organohalogen Compounds, 2008, V. 70. P. 001467-001470. |
|
| TRRD: Technology for extraction, storage, and use of knowledge about the structural-functional organization of the transcriptional regulatory regions in the eukaryotic genes N.A. Kolchanov, E.V. Ignatieva, O.A. Podkolodnaya, E.A. Ananko, T.M. Khlebodarova, I.L. Stepanenko, T.I. Merkulova, V.M. Merkulov, N.L. Podkolodnyy, A.G. Romashchenko INTELL DATA ANAL, 2008, No. 5, Vol. 12, P. 443-461 |
|
| uORFs, reinitiation and alternative translation start sites in human mRNAs Kochetov A.V., Ahmad S., Ivanisenko V., Volkova O.A., Kolchanov N.A., Sarai A. FEBS LETT, 2008, 582. 1293-1297 |
|
| Associative Network Discovery (AND) компьютерная система для автоматической реконструкции сетей ассоциативных знаний о молекулярно-генетических взаимодействиях П.С. Деменков, Е.Э. Аман, В.А. Иванисенко Вычислительные технологии, 2008, 2, 13, 15-19 |
|
| 2007 | Компьютерно-экспериментальный подход к дизайну полифункционального геносенсора, полученного на основе промотора гена yfiA Escherichia coli Тикунова Н.В., Хлебодарова Т.М., Крачко А.В., Степаненко И.Л., Колчанов Н.А. Doklady Akademii Nauk, 2007, 417, №6 , 835-839 |
| Detection of new potentially active DRE sites in regulatory region of human genes encoding components of Ah receptor cytosolic complex Nedosekina EA, Oshchepkov DY, Katokhin AV, Kuznetsova TN, Shamanina MY, Mordvinov VA, Tsyrlov I Organohalogen Compounds, 2007, V. 69. P. 1889-1892. |
|
| A mathematical model for the adenylosuccinate synthetase reaction involved in purine biosynthesis Evgeniya A Oshchepkova-Nedosekina, Vitalii A Likhoshvai THEOR BIOL MED MODEL, 2007, 4:11 |
|
| Mathematical model for suppression of subgenomic hepatitis C virus RNA replication in cell culture Mishchenko E.L., Bezmaternykh K.D., Likhoshvai V.A., Ratushny A.V., Khlebodarova T.M., Sournina N. Yu., Ivanisenko V.A., Kolchanov N.A. Journal of Bioinformatics and Computational Biology, 2007, Vol. 5, No. 02B pp.593-609 |
|
| Phylogenetic analysis of the p53 and p63/p73 gene families Pintus S.S., Fomin E.S., Oshurkov I.S., Ivanisenko V.A. In Silico Biology, 2007, 7(3), pp.319-332 |
|
| Web-based Computational Tools for the Prediction and Analysis of Post-translational Modifications of Proteins Ivanisenko V.A., Afonnikov D.A., Kolchanov N.A. Methods in Molecular Biology, 2007, 446, 363-384. |
|
| Application of bioinformatics resources for genosensor design Khlebodarova T.M., Tikunova N.V., Kachko A.V., Stepanenko I.L., Podkolodny N.L., Kolchanov N.A. Journal of Bioinformatics and Computational Biology, 2007, V.5. P.507-520 |
|
| Иммунохимические свойства белка PrM и С концевого фрагмента белка М вируса Западного Нила Богачек М.В., Протопопова Е.В., Терновой В.А., Качко А.В., Иванова А.В., Иванисенко В.А., Швалов А.Н., Локтев В.Б. Molecular Biology, 2007, 1, 41, 8-17 |
|
| 2006 | Математическая модель репликации РНК репликона вируса гепатита С в клеточной культуре. Моделирование действия потенциальных лекарственных препаратов Безматерных К.Д., Мищенко Е.Л., Лихошвай В.А., Хлебодарова Т.М.,Иванисенко В.А. BIOPHYSICS, 2006, т. 51, выпуск N7 |
| Подтверждение функциональности дополнительного Zn2+ сайта связывания в мутантной форме G245C белка p53 Э. С. Фомин, В. А. Иванисенко BIOPHYSICS, 2006, N7, т.51 |
|
| Предсказание изменения термодинамической стабильности белков при одиночных аминокислотных заменах Деменков П.С., Аман Е.Э., Иванисенко В.А. BIOPHYSICS, 2006, Т. 7, Вып. 51 |
|
| Филогенетический анализ семейства p53 Пинтус С.С., Фомин Э.С., Иванисенко В.А., Колчанов Н.А. BIOPHYSICS, 2006, т.51, вып.4, с.640-649 |
|
| Prediction of the Changes in Thermodynamic Stability of Proteins Caused by Single Amino Acid Substitutions Demenkov P.S., Aman E.E., Ivanisenko V.A. BIOPHYSICS, 2006, т. 51, выпуск N7 |
|
| Библиотека программных компонент MOLKERN для построения программ молекулярного моделирования Фомин Э.С., Алемасов Н.А., Чирцов А.С., Фомин А.Э. BIOPHYSICS, 2006, т.51, вып.7 |
|
| 2005 | Иммунохимические свойства рекомбинантных полипептидов, моделирующих домены I и II белка Е вируса Западного Нила Богачек М.В., Протопопова Е.В., Терновой В.А., Качко А.В., Иванова А.В., Иванисенко В.А., Локтев В.Б. Molecular Biology, 2005, N 5, т. 39, с. 813-822. |
| Актуальные проблемы компьютерной протеомики Иванисенко В.А., Афонников Д.А., Николаев С.В., Пинтус С.С., Крестьянова М.А., Пальянов А.Ю, Титов И.И. Информационный вестник ВОГИС, 2005, т.9, с.162-178 |
|
| 2004 | Обнаружение эмпирической аксиоматической теории в условиях шумов. Деменков П.С. Вычислительные системы, 2004 |
| 2002 | Быстрый генетический алгоритм для анализа вторичной структуры РНК Титов И.И., Воробьев Д.Г., Иванисенко В.А., Колчанов Н.А. RUSS CHEM B+, 2002, N7, c. 1047-1056 |
| Локализация антигенной детерминанты белка Е вируса клещевого энцефалита, узнаваемой антигемагглютинирующими моноклональными антителами, с помощью пептидной фаговой библиотеки Локтев А.В., Кувшинов В.Н., Меламед Н.В., Иванисенко В.А., Мишин В.П., Ильичев А.А. Вопросы вирусологии, 2002, N.2, Т.47, С.31-34 |
|
| Локализация конформационного эпитопа гликопротеина gp120 ВИЧ-1, узнаваемого вируснейтрализующими моноклональными антителами 2G12 Туманова О.Ю., Кувшинов В.Н., Ильичев А.А., Некрасов Б.Г., Иванисенко В.А., Козлов А.П., Сандахчиев Л.С. Molecular Biology, 2002, N.4, Т.36, С.657-663 |
|
| 2000 | Анализ чужеродных эпитопов, встроенных в НВсАg. Возможные пути решения проблемы самоорганизации химерных коровых частиц Карпенко Л.И., Иванисенко В.А., Пика И.С., Чикаев Н.А., Ерошкин А.М., Меламед Н.В., Веремейко Т.А., Ильичев А.А. Molecular Biology, 2000, N.2, Т.34, С.223-229 |
| 1997 | Поиск сайтов, содержащих функционально важные замены в наборах родственных или мутантных белков Иванисенко В.А., Ерошкин А.М. Molecular Biology, 1997, N.5, Т.31, С.880-887. |
| Использование фаговой библиотеки пептидов в картировании группоспецифического гемагглютинирующего домена гликопротеина Е2 альфа-вирусов Кузьмичева Г.А., Кувшинов В.Н., Разумов И.А., Иванисенко В.А., Ерошкин А.М., Мишин В.П., Ушакова Т.А., Локтев В.Б., Ильичев А.А. MOL GENET MICROBIOL+, 1997, N.4, С.25-29 |
|
| Экспрессия фрагментов гена Е вируса японского энцефалита в клетках Escherichia coli Белавин П.А., Нетесова Н.А., Решетников С.С., Иванисенко В.А., Ерошкин А.М., Протопопова Е.В., Локтев В.Б., Малыгин Э.Г. Биотехнология, 1997, N.3, С.3-10 |
 Конференции
Конференции
| 2011 | Analysis of protein functional sites encoding features Медведева И.В., Деменков П.С., Иванисенко В.А. International German/Russian Summer School on Integrative Biological Pathway Analysis and Simulation |
| Analysis of protein functional sites encoding features Медведева Ирина Вадимовна, Деменков Павел Сергеевич, Иванисенко Владимир Александрович ISMB/ECCB 2011 |
|
| Моделирование генных сетей: система GeneNet в 2011 году. Тимонов В.С., Гунбин К.В., Казанцев Ф.В., Акбердин И.Р., Деменков П.С., Лашин С.А., Ананько Е.А., Иванисенко В.А., Подколодный Н.Л., Колчанов Н.А. Международная конференция "Современные проблемы математики, информатики и биоинформатики" |
|
| The visual reconstruction and analysis of biomolecular systems: The GeneNet software in 2011 Vladimir Timonov, Fedor Kazantsev, Konstantin Gunbin, Ilya Akberdin, Pavel Demenkov, Elena Ananko, Nikolay Podkolodny, Vladimir Ivanisenko, Nikolay Kolchanov International German/Russian Summer School on Integrative Biological Pathway Analysis and Simulation |
|
| A mathematical model for the suppression of subgenomic Hepatitis C virus replication in Huh-7 cells in the presence of the NS3 protease inhibitors Мищенко Е., Иванисенко Н., Акбердин И., Лихошвай В., Козлов К., Гурский В., Самсонова М., Колчанов Н., Иванисенко В. International Conference on Systems Biology (ICSB 2011), Хайдельберг, Германия, 28 августа-1 сентября |
|
| Математическая модель подавления репликации субгеномного репликона вируса гепатита С в клетке с ингибитором NS3 протеазы SCH503034 Мищенко Е., Иванисенко Н., Акбердин И., Лихошвай В., Козлов К., Гурский В., Самсонова М., Колчанов Н., Иванисенко В. Санкт-Петербургский научный форум “Наука и общество. Физиология и медицина 21 века. VI Петербургская встреча лауреатов нобелевской премии”, Санкт-Петербург, Россия, 19-23 сентября |
|
| 2010 | Microbial metagenome annotation with bioinformatics and computational systems biology methods Ivanisenko, Demenkov, Pintus, Ivanisenko, Goncharova, Kostjukova, Levitski, Selezneva, .... Kolchanov 7th International conference on bioinformatics of genome regulation and structure\system biology (BGRS/SB'20010). June 20-27, 2010, Novosibirsk |
| Modeling of the suppressive effect of NS3 protease inhibitor on HCV subgenomic replicon replication in Huh-7 cells Bezmaternykh K.D., Mishchenko E.L.,Likhoshvai V.A., Ivanisenko V.A., Kolchanov N.A. 7th International conference on bioinformatics of genome regulation and structure\system biology (BGRS/SB'20010). June 20-27, 2010, Novosibirsk |
|
| Reconstruction of molecular genetic networks: text mining as a tool for knowledge discovery V.A. Ivanisenko, P.S. Demenkov, T.V. Ivanisenko Joint Indo-Russian Workshop "Predictive Biology using Systems and Integrative analysis and Methods", November 15-19, 2010, p 8-9, Chandigarh, India |
|
| Computer Sytem SitEx for analyzing protein functional sites in eukaryotic gene structure Медведева И. В., Деменков П. С., Иванисенко В. А. The Young Scientists School "Bioinformatics and system biology", Novosibirsk, Russia, 28-29 June 2010 |
|
| Разработка базы данных химических соединений - потенциальных маркеров заболеваний Иванисенко В.А., Сайк О.В., Деменков П.С., Мошкин П.М. XVII Российский национальный конгресс "Человек и лекарство"., Москва, 16 апреля 2010 г. |
|
| Качество спермы и гормональный статус мужчин кемеровской популяции Гуторова Н.В., Осадчук Л.В., Клещёв М.А., Кузнецова Н.Н. XXI съезд физиологического общества им. И. П. Павлова, 19-25 сентября г. Калуга |
|
| Показатели мужской фертильности у жителей г. Архангельска Клещев М. А. Осадчук Л. В. Гуторова Н. В. Типисова Е. В. XXI съезд физиологического общества им. И. П. Павлова |
|
| Региональные и этнические различия в мужской фертильности и гормональном статусе у жителей Сибири Осадчук Л. В., Клещев М. А., Гуторова Н. В., Еркович А. А., Темников Н. Д., Шантанова Л. Н., Кузнецова Н. Н., Осадчук А. В. XXI съезд физиологического общества им. И. П. Павлова |
|
| Генная сеть активации макрофага: участие ксенобиотков в регуляции экспрессии генов цитокинов Ощепкова Е.А., Ощепков Д.Ю., Кашина Е.В., Антонцева Е.В., Фурман Д.П., Мордвинов В.А. Всероссийская научная конференция "Молекулярно-генетические основы функционирования цитокиновой сети в норме и при патологии", 15-17 сентября 2010г. |
|
| Интегрированная система для информационной поддержки исследования механизмов регуляции транскрипции Н.Л. Подколодный, Е.В. Игнатьева, Д.А. Рассказов, О.А. Подколодная, Е.А. Ананько, Н.Н. Подколодная, Е.М. Залевский Всероссийская научная конференция "Электронные библиотеки: перспективные методы и технологии, электронные коллекции" – RCDL’2010, Казань, Россия, 2010 |
|
| Сравнительный анализ производительности процессоров PowerXCell8i и Intel X7560 на примере задач молекулярной динамики Фомин Э.С., Н.А. Алемасов, С.А.Матвиенко Высокопроизводительные параллельные вычисления на кластерных системах HPC2010, Материалы X Международной конференции, г.Пермь, 1-3 ноября, 2010 г. |
|
| Математическое моделирвание репликации репликона вируса гепатита С: предсказание эффетивности действия новых потенциальных лекарств Мищенко Е.Л., Иванисенко Н.В., Мищенко А.М., Лихошвай В.А., Иванисенко В.А. Международная научно-практическая конференция "Высокие технологии, фундаментальные и прикладные исследования в физиологии и медицине" 23-26 ноября, 2010, г. Санкт-Петербург |
|
| Экспериментально-компьютерное исследование генов, вовлекаемых в реакцию макрофага на диоксин Ощепков Д.Ю., Ощепкова Е.А., Кашина Е.В., Антонцева Е.В., Фурман Д.П., Мордвинов В.А. Международная научно-практическая конференция "Высокие технологии, фундаментальные и прикладные исследования в физиологии и медицине" 23-26 ноября, 2010, г. Санкт-Петербург |
|
| Предварительные результаты исследования репродуктивного здоровья мужского населения Республики Бурятия Шантанова Л.Н., Башелханов И.С., Осадчук Л.В., Осадчук А.В., Клещев М.А., Гуторова Н.В. Международная научно-практическая конференция "Актуальные исследования Байкальской Азии". |
|
| Изучение основных параметров фертильности и гормонального статуса у мужчин города Новосибирска А. В. Попова, М. А. Клещев, Н. Д. Темников, Л. В. Осадчук Научная конференция "Фундаментальные науки – медицине" |
|
| Компьютерный анализ метагеномных данных: предсказание количественной величины специфической активности белков Иванисенко В.А., Деменков П.С., Пинтус С.С., Иванисенко Т.В., Гончарова Н.И., Розанов А., Брянская А., Кострюкова Е.С., Левитский С.А., Селезнева О.В., Чукин М.М., Ларин А.К., Кондратов И.Г., Лазарев В.Н., Пельтек С.Е., Говорун В.М. Постгеномные методы анализа в биологии, лабораторной и клинической медицине |
|
| Computer analysis of conformationalpeptides in protein families A.O. Bragin, P.S. Demenkov, V.A. Ivanisenko 7th International conference on bioinformatics of genome regulation and structure\system biology (BGRS/SB'20010). June 20-27, 2010, Novosibirsk |
|
| Analisys of human proteome based on protein stability to mutations P.S. Demenkov, O.A. Korepanova, T.V. Ivanisenko, V.A. Ivanisenko 7th International conference on bioinformatics of genome regulation and structure\system biology (BGRS/SB'20010). June 20-27, 2010, Novosibirsk |
|
| ITMSys: An interactive web-based text-mining system for automated analysis of the full-text articles Ivanisenko T.V., Demenkov P.S., Ivanisenko N.V., Ivanisenko V.A. 7th International conference on bioinformatics of genome regulation and structure\system biology (BGRS/SB'20010). June 20-27, 2010, Novosibirsk |
|
| ANDCell and ANDNanobiotech: Associative network discovery systems in systems biology and nanobiotechnology V.A. Ivanisenko, P.S. Demenkov, T.V. Ivanisenko, N.L. Podkolodny 7th International conference on bioinformatics of genome regulation and structure\system biology (BGRS/SB'20010). June 20-27, 2010, Novosibirsk |
|
| Computer system SitEx for analyzing protein functional sites in eukaryotic gene structure I.V. Medvedeva, P.S. Demenkov, V.A. Ivanisenko 7th International conference on bioinformatics of genome regulation and structure\system biology (BGRS/SB'20010). June 20-27, 2010, Novosibirsk |
|
| The nanobiotechnology database V.A. Ivanisenko, N.L. Podkolodnyy, P.S. Demenkov, T.V. Ivanisenko, N.N. Podkolodnaya, T.M. Khlebodarova, E.V. Ignatieva, O.A. Podkolodnaya, E.A. Ananko, and N.A. Kolchanov 7th International conference on bioinformatics of genome regulation and structure\system biology (BGRS/SB'2010). June 20-27, 2010, Novosibirsk |
|
| A computer analysis of functional relationships between genes associated with multifactorial diseases: pre-eclampsia as an example E. Tiys, P. Demenkov, E. Vashukova, A. Glotov, V. Ivanisenko 7th International conference on bioinformatics of genome regulation and structure\system biology (BGRS/SB'20010). June 20-27, 2010, Novosibirsk |
|
| 2009 | модификация фермента ксилозоизомеразы E.Coli дисульфидным мостиком для повышения стабильности Розанов А.С., Цырульников А.О., Иванисенко В.А., Пельтек С.Е. 13 - международная школа конференция молодых ученых, 28 сентября - 2 октября. Биология наука 21-го века |
| Associative network discovery systems in systems biology and nanobiotechnology Ivanisenko V.A. International Autumn School for Young Scientists on Computational Systems Biology and Bioinformatics |
|
| Suppressive effect of potencial individual drugs or their combinations on HCV replication in cells: a mathematical model Mishchenko E.L. International Autumn School for Young Scientists on Computational Systems Biology and Bioinformatics |
|
| Автоматическая экстракция знаний из научных публикаций в области системной биологии и нанобиотехнологии Иванисенко В.А., Деменков П.С., Иванисенко Т.В., Подколодный Н.Л., Колчанов Н.А. IV Петербургская встреча Нобелевских лауреатов. С.-Петербург. 21-25 сентября 2009г |
|
| ANDCell and ANDNanobiotech: associative network discovery systems in systems biology and nanobiotechnology Vladimir A. Ivanisenko, P.S. Demenkov, T.V. Ivanisenko Proceedings of the German-Russian Forum Biotechnology GRFB’09, 2009 |
|
| Использование архитектуры Cell/B.E. для ускорения выполнения пакета молекулярного моделирования MOLKERN Алемасов Н.А. XVI Международная конференция студентов, аспирантов и молодых ученых "Ломоносов", Москва, 14-17 апреля 2009 |
|
| Алгоритм расчёта ближних невалентных взаимодействий на процессоре PowerXCell8i. SPE-центричная модель Алемасов Н.А., Фомин Э.С. Девятая международная конференция-семинар "Высокопроизводительные параллельные вычисления на кластерных системах", Владимирский государственный университет им. А.Г. и Н.Г. Столетовых, Владимир, 2–3 ноября 2009 года |
|
| Нанобиотехнология: извлечение знаний методами text-mining Колчанов Н.А., Иванисенко В.А., Деменков П.С., Подколодный Н.Л. Конференция "Химическая биология – Фундаментальные проблемы бионанотехнологии", 10 – 14 июня 2009 г., Новосибирск |
|
| Изучение влияния на термостабильность ксилозоизомераза e.coli введения межъсубеденичного дисульфидного мостика Розанов А.С., Цырульников А.О., Иванисенко В.А., Пельтек С.Е. Конференция IV Международная конференция "Биология: от молекулы до биосферы" 17 - 21 ноября была перенесена |
|
| Информационная система ANDCell-ANDVisio для построения ассоциативных сетей в области молекулярной биологии Деменков П. С., Яркова Е. Э., Иванисенко В. А. Седьмая международная конференция памяти академика А.П. Ершова. Рабочий семинар "Наукоемкое программное обеспечение" 15-19 июня 2009. Новосибирск |
|
| Нанобиотехнология: извлечение знаний методами text-mining Иванисенко В.А., Деменков П. С., Подколодный Н.Л., Яркова Е. Э., Иванисенко Т. В., Хлебодарова Т.М., Колчанов Н.А. Пятый московский международный конгресс “Биотехнология: состояние и перспективы развития” |
|
| 2008 | Data mining and network construction Ivanisenko V.A. 11th Meeting of the German/Russian Virtual Network of Bioinformatics for "Computational Systems Biology". August 25-26, 2008. Bielefeld, Germany |
| A mathematical model for the suppressive effect of subgenomic hepatitis c virus replication in cells in the presence of potential individual drugs or their combinations E.L. Mishchenko, K.D. Bezmaternykh, V.A. Likhoshvai, V.A. Ivanisenko, N.A. Kolchanov. 6th international conference on bioinformatics of genome regulation and structure BGRS’2008, Novosibirsk, June 22-28 |
|
| Andcell: a computer system for automated extraction of knowledge about molecular genetic interactions and regulations from pubmed abstracts and their representation as semantic association networks Vladimir A. Ivanisenko, P.S. Demenkov, T.V. Ivanisenko 6th international conference on bioinformatics of genome regulation and structure BGRS’2008, Novosibirsk, June 22-28 |
|
| Coevolution of protein domains of P53 and MDM2 – key proteins of apoptosis S.S. Pintus, V.A. Ivanisenko 6th international conference on bioinformatics of genome regulation and structure BGRS’2008, Novosibirsk, June 22-28 |
|
| Estimation of minimal drug treatment duration for clearance of an Huh-7 cell from hepatitis C virus replicon based on mathematical modeling Mishchenko E.L., Bezmaternykh K.D., Likhoshvai V.A., Ivanisenko V.A., Kolchanov N.A. 6th international conference on bioinformatics of genome regulation and structure BGRS’2008, Novosibirsk, June 22-28 |
|
| Modeling of drug effects on hepatitis C virus replication in an Huh-7 cell Mishchenko E.L., Bezmaternykh K.D., Likhoshvai V.A., Ivanisenko V.A., Kolchanov N.A. 6th international conference on bioinformatics of genome regulation and structure BGRS’2008, Novosibirsk, June 22-28 |
|
| Nanobiotechnology and its application to biomedicine: text-mining knowledge extraction and integration Ivavisenko V.A. Demenkov P.S., Yarkova E.E., Ivavisenko N.V., Ivavisenko T.V. Sournina N.Yu., Podkolodny N.L., Khlebodarova T.M., Ibragimova S.S., Smirnova O.G. 6th international conference on bioinformatics of genome regulation and structure BGRS’2008, Novosibirsk, June 22-28 |
|
| PDBSite database and PDBSiteScan tool: template-based docking and recognition of functional sites in protein 3d structure Ivanisenko V.A., Ivanisenko N.V., Ivanisenko T.V. 6th international conference on bioinformatics of genome regulation and structure BGRS’2008, Novosibirsk, June 22-28 |
|
| Protein connectivity in molecular-genetic networks depends on protein susceptibility to single mutations Demenkov P.S., Yarkova E.E., Ivanisenko V.A., Ivanisenko T.V. 6th international conference on bioinformatics of genome regulation and structure BGRS’2008, Novosibirsk, June 22-28 |
|
| Protein Functional Sites Projection on Exon Structure Of Gene for ATP-, ADP-, AMP-binding Sites I.V. Medvedeva, V.A. Ivanisenko International Autumn School for Young Scientists on Computational Systems Biology and Bioinformatics |
|
| Protein Functional Sites Ivanisenko V.A. International Autumn School for Young Scientists on Computational Systems Biology and Bioinformatics |
|
| Эффект потенциальных лекарственных препаратов на репликацию субгеномного репликона вируса гепатита C в клетке: математическая модель Е.Л. Мищенко, К.Д.Безматерных, В.А. Лихошвай, В.А. Иванисенко IV Международная конференция "Фундаментальные науки - медицине", г. Новосибирск, 2008 г. |
|
| Математическое моделирование репликации репликона ВГС в клетке. Исследование возможных клеточных факторов Безматерных К.Д., Мищенко Е.Л., Иванисенко В.А., Лихошвай В.А IV съезд микробиологов Узбекистана, тезисы докладов, г. Ташкент, 9-10 октября 2008 |
|
| Информационные ресурсы для дизайна новых геносенсоров. разработка полифункциональных геносенсоров на основе промоторов генов yfiA и dps Escherichia coli Т. М. Хлебодарова, А.В. Качко, Н.В. Тикунова, В.А. Лихошвай, Т.Ю.Степанова, Д.Ю. Ощепков, И.Л. Степаненко, Ю.Г. Матушкин IV съезд Российского Общества биохимиков и молекулярных биологов, 10-15 мая, Новосибирск, 2008 |
|
| Drug targets discovery with text-mining and bioinformatics approaches: ANDCell and PDBSiteScan tools Ivanisenko V.A., Demenkov P.S., Yarkova E.E. Joint Indo-Russian Workshop "Systems Biology and Genome Informatics of M. tuberculosis and other infectious diseases" October 12-14, 2008. Novosibirsk |
|
| ANDCell: a tool for automated knowledge extraction from PubMed and reconstruction of association networks Ivanisenko V.A., Yarkova E.E., Demenkov P.S., Kochetov A.V. Proceedings of the 5th International Symposium on Integrative Bioinformatics. P 327, 20-22 August 2008, Germany |
|
| Knowledge discovery for biotechnology, biomedicine and nanobioengineering using text- and data-mining Ivanisenko V.A., Demenkov P.S., Yarkova E.E. Russian-Polich Symposium "Perspectives of post-genomic biotechnology" in the frames of jubilee celebrations on 50 –anniversary of cooperation between the Russian and Polish Academies of Sciences, October 15-17, 2008, Moscow |
|
| Параллельная реализация программного комплекса молекулярного моделирования "MOLKERN" с использованием OpenMP Алемасов Николай Александрович Технологии Microsoft в теории и практике программирования, Конференция-конкурс работ студентов, аспирантов и молодых ученых, Новосибирск, Академгородок |
|
| Protein connectivity in molecular-genetic networks depends on protein susceptibility to single mutations Demenkov P.S., Yarkova E.E., Ivanisenko T.V., Ivanisenko V.A. 6th international conference on bioinformatics of genome regulation and structure BGRS’2008, Novosibirsk, June 22-28 |
|
| 2007 | Analysis of Distribution of Protein Functional Sites on Gene Structure Medvedeva I. V., Demenkov P. S., Ivanisenko V. A., and Kolchanov N. A. 2007 International Conference on Bioinformatics and Computational Biology (BIOCOMP'07: June 25-28, 2007) |
| Associative network and protein structure discovery: a software complex for facilitating search of targets for drugs, drug design, and evaluation of molecular toxicity Ivanisenko V.A., Demenkov P.S., Aman E.E., S.S. Pintus S.S., Kolchanov N.A. 3-rd International Conference "Basic Science for Medicine", September 2-8, 2007, Novosibirsk, Russia |
|
| Signals influencing general translational efficiency of eukaryotic mRNAs Ivanisenko VA, Mischenko EL, Kochetov A.V., Ivanisenko V.A., Titov I.I., Kolchanov N.A., Sarai A. 3-rd Moscow Conference on Computational Molecular Biology "MCCMB' 2007". July 27-31, 2007, Moscow, Russia |
|
| Associative network discovery (AND) – Software package for automated reconstruction of molecular-genetic association networks Aman E.E., Demenkov P.S., Nemiatov A., Ivanisenko V.A. 3-rd Moscow Conference on Computational Molecular Biology "MCCMB' 2007". July 27-31, 2007, Moscow, Russia |
|
| Prediction in changes of protein thermodynamic stability upon single mutations P.S. Demenkov 8th Meeting German / Russian Virtual Network on Computational Systems Biology 5th and 6th November 2007 |
|
| Математическая модель репликации РНК репликона вируса гепатита С в клетке. Построение и варианты использования Безматерных К.Д., Мищенко Е.Л., Иванисенко В.А., Лихошвай В.А., Колчанов Н.А. III Международная конференция "Фундаментальные науки - медицине", г. Новосибирск, 2-8 сентября 2007 г. |
|
| Mathematical Modeling of the HCV Drugs Combinations Effect Bezmaternykh K.D., Mishchenko E.L., Ivanisenko V.A., Likhoshvai V.A. In Proceedings of the 3-rd Moscow Conference on Computational Molecular Biology. Moscow, Russia, July 27-31, 2007 |
|
| Bioinformatics and its application in addressing the chalanges of biotechnology and medicine Ivanisenko V.A. International Conference "Scenarios for a co-ordinated approach to sustainable S&T co-operation with the eastern neighbours of the EU", Moscow 3-4 December, 2007 |
|
| Параллельная реализация задач молекулярного докинга и виртуального скрининга Алемасов Николай Александрович, Фомин Эдуард Станиславович IV Российская-германская школа по параллельным вычислениям на высокопроизводительных вычислительных системах, Новосибирск |
|
| Structure discovery – computer tools for protein analysis and search of drug target V.A. Ivanisenko, P.S. Demenkov, E.E. Aman, S.S. Pintus, E.S. Fomin. The Fifth Moscow International Congress "Biotechnology: State of the Art and Prospects of Development" 14-18 марта 2007 года |
|
| Textomics: the instrument for biological knowledge discovery E.E. Aman, P.S. Demenkov, V.A. Ivanisenko The Fifth Moscow International Congress "Biotechnology: State of the Art and Prospects of Development" 14-18 марта 2007 года |
|
| Associative Network Discovery (AND) – компьютерная система для автоматической реконструкции ассоциативных сетей молекулярно-генетических взаимодействий Aman E.E., Demenkov P.S., Nemiatov A., Ivanisenko V.A. VI Всероссийской научно – практической конференции AS’2007 (СИСТЕМЫ АВТОМАТИЗАЦИИ в образовании, науке и производстве) |
|
| Анализ картирования функциональных сайтов белков на экзонной структуре гена Медведева И. В. XLV международная научная студенческая конференция: Студент и научно-технический прогресс, Новосибирск 10-12 апреля 2007г. |
|
| Математическая модель репликации репликона вируса гепатита С в клетках Huh-7 Безматерных К.Д., Мищенко Е.Л., Иванисенко В.А., Лихошвай В.А. XLV международная научная студенческая конференция: Студент и научно-технический прогресс, Новосибирск 10-12 апреля 2007г. |
|
| Математическое моделирование комбинационного действия лекарственных препаратов против вируса гепатита С. Безматерных К.Д., Мищенко Е.Л., Иванисенко В.А., Лихошвай В.А. Международная молодежная научно-методическая конференция "Проблемы молекулярной и клеточной биологии", г. Томск |
|
| 2006 | Bioinformatics and mechanisms of molecular pathology N.A. Kolchanov, V. Ivanisenko, M. Ponomarenko, E. Aman, E. Ignatieva, T. Kuznetsova, V. Mordvinov, P. Demenkov, A. Ratushny, V.Likhoshvai, E. Ananko, N. Podkolodny, N. Podkolodnaya, A.. Romaschenko 3rd International conference "Genomics, Proteomics, Bioinformatics and Nanotechnologies for Medicine" July 12-16, 2006, Novosibirsk, Russia |
| Hepatitis C virus: application of gene networks technology to searching for perspective pharmacological targets Ivanisenko VA, Mischenko EL, Bezmaternikh KD, Ratushny AV, Likhoshvai VA,Titov II, Korotkov RO, Khlebodarova TM, Kolchanov NA. 3rd International conference "Genomics, Proteomics, Bioinformatics and Nanotechnologies for Medicine" July 12-16, 2006, Novosibirsk, Russia |
|
| Phylogenetic analysis of the p53 and p63/p73 gene families Pintus SS, Ivanisenko VA 3rd International conference "Genomics, Proteomics, Bioinformatics and Nanotechnologies for Medicine" July 12-16, 2006, Novosibirsk, Russia |
|
| New Zn2+ binding sites in the p53 mutants are promising targets for antitumor drug design Fomin E.S., Oshurkov I.S., Ivanisenko V.A. 3rd International conference "Genomics, Proteomics, Bioinformatics and Nanotechnologies for Medicine" July 12-16, 2006, Novosibirsk, Russia |
|
| Analysis of the tertiary structure of the PPAR and RXR transcriptional factors and their mutant variants Aman E.E., Demenkov P.S., Ivanisenko V.A. 5th International Conference on Bioinformatics of Genome Regulation and Structure (BGRS'2006), Novosibirsk, Russia (July 14-22) |
|
| Database GenSensor as informational source for design of biosensors. Experimental development of biosensor based on yfiA gene Khlebodarova T.M., Kachko A.V., Stepanenko I.L., Tikunova N.V. 5th International Conference on Bioinformatics of Genome Regulation and Structure (BGRS'2006), Novosibirsk, Russia (July 14-22) |
|
| Development of a computer system for the automated reconstruction of molecular-genetic interaction networks Aman E.E., Demenkov P.S., Pintus S.S., Nemiatov A.I., Apasieva N.V., Dubovenko E.A., Ignatieva E.V., Podkolodny N.L., Ivanisenko 5th International Conference on Bioinformatics of Genome Regulation and Structure (BGRS'2006), Novosibirsk, Russia (July 14-22) |
|
| FASTPROT: A computtational workbench for analysis of function, activity and structure of protein Ivanisenko V.A., Demenkov P.S., Fomin E.S., Oshurkov I.S., Korotkov R.O., Aman E.E., Pintus S.S., Startcev K.S., Tatarnikova L.Yu., Panina I.I., Sobolev A.A., Selenev I.V., Elohov V.J., Golovina A.N., Aknazarov Z.I., Sharonova I.V., Krestianova M.A., etc. 5th International Conference on Bioinformatics of Genome Regulation and Structure (BGRS'2006), Novosibirsk, Russia (July 14-22) |
|
| HCV-KINET database: kinetic parameter reactions and regulatory processes of the life cycle of the hepatitis C virus Mishchenko E.L., Korotkov R.О., Ivanisenko V.A. 5th International Conference on Bioinformatics of Genome Regulation and Structure (BGRS'2006), Novosibirsk, Russia (July 14-22) |
|
| PDBSite database and PDBSiteScan tool: recognition of functional sites in protein 3D structure and template-based docking Ivanisenko V.A., Ivanisenko T.V., Sharonova I.V., Krestyanova M.A., Ivanisenko N.V., Grigorovich D.A. 5th International Conference on Bioinformatics of Genome Regulation and Structure (BGRS'2006), Novosibirsk, Russia (July 14-22) |
|
| Prediction in changes of protein thermodynamic stability upon single mutations Demenkov P.S, Ivanisenko V.A. 5th International Conference on Bioinformatics of Genome Regulation and Structure (BGRS'2006), Novosibirsk, Russia (July 14-22) |
|
| Rise of new zn2+ binding sites can be a molecular mechanism for impaired function of the p53 mutants Fomin E.S., Oshurkov I.S., Ivanisenko V.A. 5th International Conference on Bioinformatics of Genome Regulation and Structure (BGRS'2006), Novosibirsk, Russia (July 14-22) |
|
| MGSmodeller - компьютерная система для конструирования, расчета и анализа моделей молекулярно-генетических систем Казанцев Ф.В. , Подколодная Н.Н., Акбердин И.Р., Безматерных К.Д., Крокус И.В., Лашин С.А., Ратушный А.В., Лихошвай В.А. 7 Всероссийская конференция молодых ученых по математическому моделированию и информационным технологиям, Красноярск, Россия, (Ноябрь 1-3) |
|
| System computer biology in Institute of Cytology of SB RAS Yu. Matushkin, E. Ananko, D. Afonnikov, V. Golomolzin, V. Ivanisenko, E. Ignat’eva, A. Katokhin, V.Likhoshvai, D. Miginskii, N. Omelyanchuk, S. Peltek, T. Khlebodarova, N. Kolchanov Saint-Petersburg International Workshop on NanoBiotechnologies |
|
| A minimum mathematical model for suppression HCV RNA replication in cell culture Безматерных К.Д., Мищенко Е.Л., Ратушный А.В., Лихошвай В.А., Хлебодарова Т.М., Иванисенко В.А. The 2d All-Russian Conference "Infocommunication and Computing Technologies and Systems", Ulan-Ude |
|
| Mathematical model for suppression of hepatitis C virus RNA replicon replication in cell culture Bezmaternikh K.D., Mishchenko E.L., Ratushny A.V., Likhoshvai V.A., Khlebodarova T.M., Ivanisenko V.A. The VII All-Russian young scientists conference on mathematical modeling and informatics technology (with foreign participants), Krasnoyarsk |
|
| Анализ распределения просайтов функциональных сайтов в пространственных структурах белков Шаронова (Медведева) И. В. XLIV Международная научная студенческая конференция "Студент и научно-технический прогресс" |
|
| 2005 | Discrete Modeling of Structure Preserving Mutations in Proteins Pavel S. Demenkov, Evgeny Y. Kharlamov 9-th Asian Logic Conference |
| 2004 | PDBSITE, PDBLIGAND and PDBSITESCAN: a computational workbench for the recognition of the structural and functional determinants in protein tertiary structures combined with protein draft docking Ivanisenko V.A., Pintus S.S., Krestyanova M.A., Demenkov P.S., Znobisheva E.K., Ivanov E.E., Grigorovich D.A. 2. Fourth Intern. Conf. on Bioinformatics of Genome Regulation and Structure |
| Разработка логико-вероятностной модели белков, определяющей структурную ограниченность белков Деменков П.С. 42-я МНСК «Студент и научно-технический прогресс» |
|
| 2003 | Emperical Theory Discovery. Деменков П.С., Витяев Е.Е. Вероятностные Идеи в Науке и Философии |
Гранты
| 2011 | Компьютерный анализ и моделирование процессов развития апикальной меристемы побега РФФИ, номер гранта 11-04-01748 |
Научное руководство
| 2011 | Предсказание изменения термодинамической стабильности белков при одиночных мутациях аминокислот Корепанова Ольга Александровна 2011-06-16 |
Патенты
| 2010 | Программа для классификации биологических текстов на английском языке (БиоТекстКласс) / The program for classification of biological texts in English (BioTextClass) Иванисенко В.А., Деменков П.С., Иванисенко Т.В. Номер патента 2010617828 |
| База данных нанобиоматериалов (НАНОБИОБАЗА)/Nanobiomaterial database (NANOBIOBASE) Иванисенко В.А., Деменков П.С., Иванисенко Т.В., Подколодный Н.Л., Хлебодарова Т.М., Игнатьева Е.В., Ананько Е.А., Подколодная О.А., Колчанов Н.А. Номер патента 2010620013 |
|
| База данных нанобиотехнологий (НАНОБИОТЕХБАЗЕ)/Nanobiotechnology database (NANOBIOTECHBASE) Иванисенко В.А., Деменков П.С., Иванисенко Т.В., Подколодный Н.Л., Хлебодарова Т.М., Игнатьева Е.В., Ананько Е.А., Подколодная О.А., Колчанов Н.А. Номер патента 2010620014 |
|
| 2008 | База данных ассоциативных сетей (ЭНДЦелл)/Associative network database (ANDCell) Иванисенко В.А., Деменков П.С., Яркова Е.Э. Номер патента 2008620439 |
| Программа для реконструкции, визуализации и анализа ассоциативных сетей (ЭНДВизио)/Program for reconstruction, visualization and analyze associative networks (ANDVisio) Иванисенко В.А., Деменков П.С., Яркова Е.Э. Номер патента 2008615929 |
|
| Компьютерная система для конструирования, расчета и анализа моделей молекулярно-генетических систем (МГСмоделлер)/ A computer system for reconsnruction/ Calculation and analysis matimatical models of molecular genetic system (MGSmodeller) Лихошвай В.А., Казанцев Ф.В., Акбердин И.Р., Безматерных К.Д. Лашин С.А., Подколодная Н.Н., Ратушный А.В Номер патента №2008612820 |
| © 2010-2026 ИЦиГ СО РАН. Все права защищены. |